
Overview
Understanding how changes in DNA impact genomic activity and affect the complex machinery of gene regulation has been a grand challenge in genomics. Google DeepMind’s AlphaGenome represents a major step forward: by taking in DNA sequences of up to one million base pairs, it predicts at single base-pair resolution hundreds of genomic tracks across diverse cell types and conditions. With both the model code and pretrained weights publicly released, the genomics community now has a powerful foundation to build on.
The original AlphaGenome model is implemented in JAX, a high-performance framework for numerical computing and deep learning. Here we present a port of AlphaGenome to PyTorch. We strived to make our implementation an accessible, readable, and hackable port of the model for the wider community to build on and adapt for their unique use cases. We also enable workflows to finetune the model on new datasets and cell types using custom data, and offer an early version of finetuning functionality in this release.
Getting Started
Using AlphaGenome in PyTorch is straightforward. The package can be installed from PyPI as:
pip install alphagenome-pytorchHere we show how to load the model from weights hosted on Hugging Face, and then run inference on a DNA sequence:
from alphagenome_pytorch import AlphaGenome
from alphagenome_pytorch.utils.sequence import sequence_to_onehot_tensor
from huggingface_hub import hf_hub_download
import pyfaidx
hf_hub_download(
"gtca/alphagenome_pytorch",
"model_all_folds.safetensors",
local_dir="."
)
model = AlphaGenome.from_pretrained("model_all_folds.safetensors")
with pyfaidx.Fasta("hg38.fa") as genome:
sequence = str(genome["chr1"][1_000_000:1_131_072])
dna_onehot = sequence_to_onehot_tensor(sequence).unsqueeze(0)
preds = model.predict(dna_onehot, organism_index=0) # 0=human, 1=mouse
# Access predictions (batch, sequence, tracks) by head name and resolution:
# - preds['atac'][1]: 1bp resolution, shape (1, 131072, 256)
# - preds['atac'][128]: 128bp resolution, shape (1, 1024, 256)The model accepts sequences of up to 1,048,576 base pairs (1 Mb) and returns predictions at single nucleotide and 128 bp resolutions for the genomic tracks it was trained on.
Numerical Equivalence with JAX
Our PyTorch port is numerically on par with the original implementation in JAX.
Small implementation differences can silently change scientific conclusions. Hence we strived to ensure our implementation has close numerical equivalence with the JAX model. We added tests for numerical equivalence of the outputs of individual model heads and full forward and backward passes through the model, gradients, and loss values.
We verified equivalence at multiple levels:
- Layer-by-layer outputs: Each convolutional block, attention mechanism, and transformer layer produces outputs within numerical precision (less than
1e-5relative error) of the JAX implementation - Full forward pass: End-to-end predictions across all genomic tracks match within floating-point precision
- Gradient computations: Backpropagation yields equivalent gradients, ensuring training dynamics remain faithful to the original implementation
- Loss values: Multinomial loss computes identically on the same inputs
We converted the pretrained weights directly from the released checkpoints so that it is easy to start working with the model with a single from_pretrained() call.
To demonstrate parity of our implementation with DeepMind’s model, we show predicted tracks in the HepG2 cell line from a 1 Mb held-out region of chromosome 19, examined in Figure 2a of the original paper:

Predicted contact maps for this region also show high concordance when comparing the two implementations (Pearson r = 0.9999):

What Can You Do With This?
Beyond drop-in replacement for the JAX implementation, our PyTorch version opens up several possibilities:
- Integration with PyTorch Ecosystems: Seamlessly combine AlphaGenome with other PyTorch-based tools and frameworks.
- Variant Effect Prediction: Compute the impact of genetic variants by running inference on reference and alternate sequences, and then compare the predicted genomic tracks. This is particularly powerful for understanding disease-associated variants.
- In Silico Mutagenesis (ISM): Systematically mutate sequences to identify important regulatory elements and understand sequence grammar.
- Finetuning on Custom Data: Perhaps most excitingly, you can adapt the model to your specific cell types, conditions, and even different species. We provide utilities for finetuning with your own genomic assay data.
In an upcoming post, we’ll dive deeper into different finetuning strategies implemented in the package, including data preparation, training best practices, and evaluation metrics to ensure that your adapted model performs well for specific use cases.
Predicting Variant Effects and In Silico Mutagenesis
Variant effect prediction is a key utility of AlphaGenome that enables interrogation of variants and their impact on disease states. We expose a simple API for predicting variant effects:
from alphagenome_pytorch.variant_scoring import VariantScoringModel
# Using previously defined AlphaGenome object
scoring_model = VariantScoringModel(
model,
fasta_path=FASTA_PATH,
gtf_path=GTF_PATH,
polya_path=POLYA_PATH,
default_organism='human',
)
ref_outputs, alt_outputs = scoring_model.predict_variant(interval, variant, to_cpu=True)To demonstrate functionality around variant analysis, we give an example of variant effect prediction and ISM for a variant impacting the TAL1 gene, examined in Figure 6 of the original paper. This variant is an oncogenic insertion impacting CD34+ common myeloid progenitors (the closest cell line in the model’s training set to the T-ALL cell of origin). We show that alphagenome-pytorch recapitulates the difference between predicted tracks of the alternate and reference sequences for the variant with high accuracy:

Our port also correctly reproduces in silico mutagenesis (ISM). We implement a simple API for running ISM:
from alphagenome_pytorch.variant_scoring import (
CenterMaskScorer,
AggregationType,
GeneMaskLFCScorer
)
# Using previously defined AlphaGenome and VariantScoringModel objects
ism_scorers = [
CenterMaskScorer(OutputType.DNASE, 501, AggregationType.DIFF_LOG2_SUM),
CenterMaskScorer(OutputType.CHIP_HISTONE, 2001, AggregationType.DIFF_LOG2_SUM),
GeneMaskLFCScorer(OutputType.RNA_SEQ),
]
print('Running ISM...')
ism_result = scoring_model.score_ism_variants(
interval=interval,
center_position=variant.position,
scorers=ism_scorers,
window_size=WINDOW_SIZE,
to_cpu=True,
progress=True,
)Results from running ISM at this locus show close agreement when compared against ISM computed using the original JAX checkpoint:
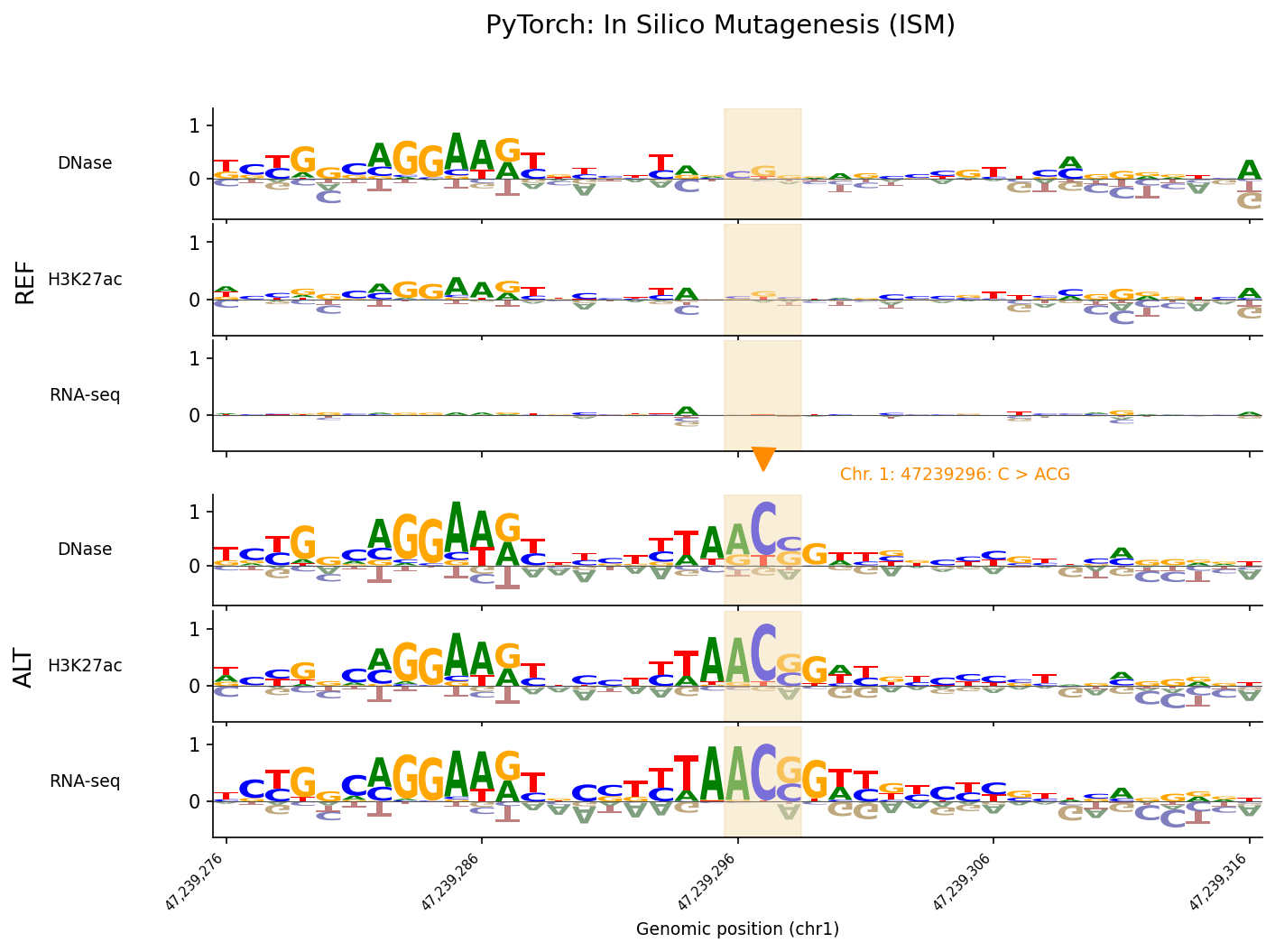
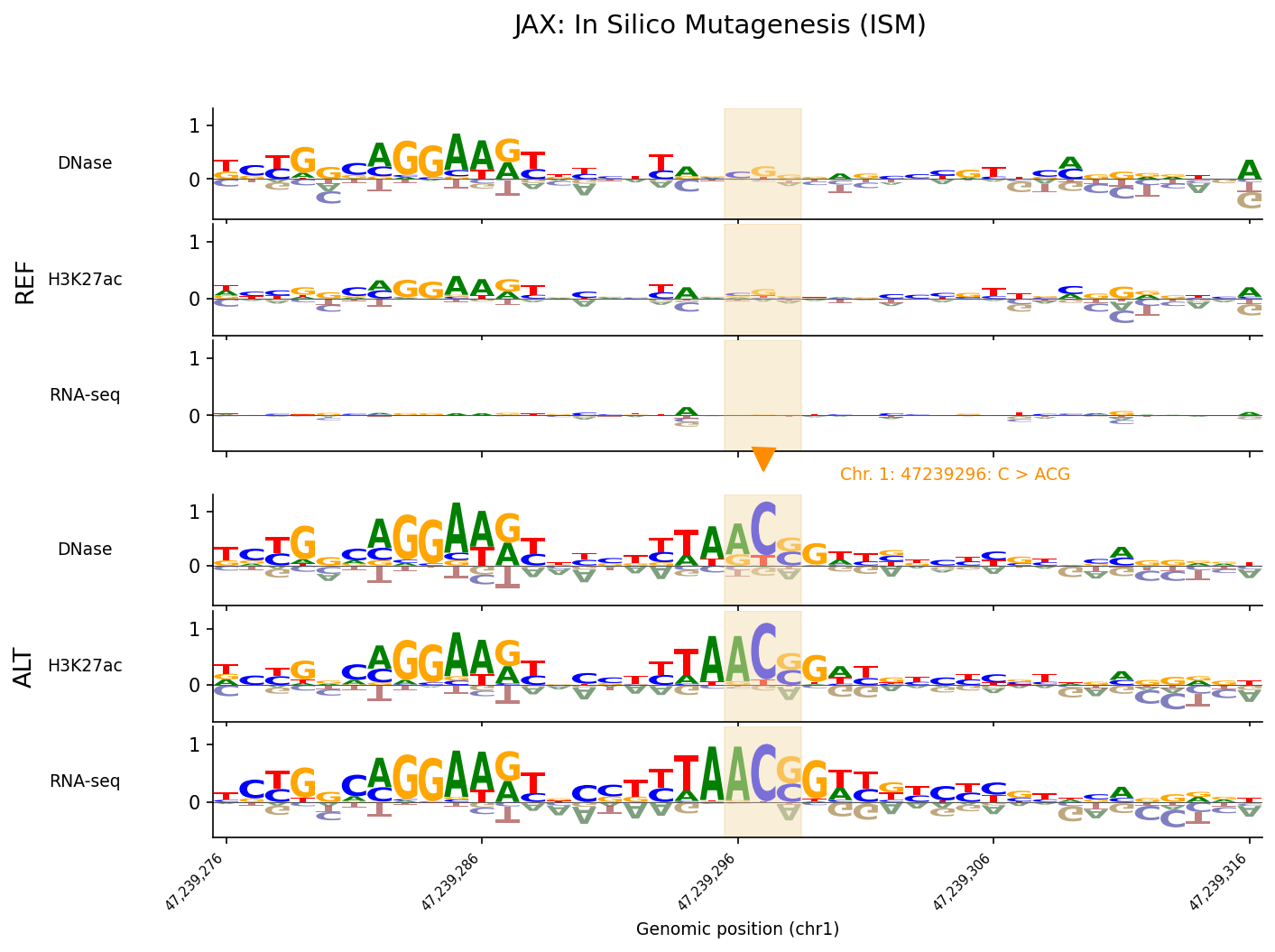
Code and Tutorials
The code is available on GitHub with detailed documentation and example notebooks to help you get started with this implementation. This is naturally a work in progress: we’re actively developing new features, improving code and performance, and working on new examples. We welcome contributions, feedback, bug reports, and stories of how this implementation has helped your research!
License
This project is a port of the google-deepmind/alphagenome_research repository licensed under the Apache License, Version 2.0:
Copyright 2026 Google LLC
The model parameters, output, and any derivatives thereof remain subject to Google DeepMind’s AlphaGenome Model Terms.
This port is licensed under the Apache License, Version 2.0 (Apache 2.0):
Copyright 2026 Danila Bredikhin, Martin Kjellberg, Christopher Zou, Alejandro Buendia, Xinming Tu, Anshul Kundaje
Licensed under the Apache License, Version 2.0 (the “License”); you may not use this except in compliance with the License. Unless required by applicable law or agreed to in writing, software distributed under the License is distributed on an “AS IS” BASIS, WITHOUT WARRANTIES OR CONDITIONS OF ANY KIND, either express or implied. See the License for the specific language governing permissions and limitations under the License.
Acknowledgements
We thank Peter Koo, Alan Murphy, Masayuki Nagai, and other members of the Koo Lab for helping initiate the GenomicsxAI organization and reviewing this post. We acknowledge Phil Wang, Miquel Anglada-Girotto, and Xinming Tu as developers of an older AlphaGenome PyTorch implementation unrelated to this port.
References
- Avsec, Ž. et al. Advancing regulatory variant effect prediction with AlphaGenome. Nature 649 (2026).
Comments
Add your reaction or comment below.